Qu'est-ce qu'un angiome ?
Le terme angiome est généralement utilisé pour désigner une anomalie localisée des vaisseaux sanguins, visible sur la peau ou au sein d’un organe. En réalité, ce terme est assez imprécis, car il regroupe des maladies très différentes. Aujourd’hui, on préfère parler d’anomalies vasculaires superficielles, qui sont classées en deux grands groupes bien distincts, notamment selon la façon dont les lésions vasculaires se forment.
- Le premier groupe correspond aux tumeurs vasculaires bénignes. Dans ce cas, il existe une multiplication anormale, appelée prolifération, des vaisseaux sanguins. Ce groupe comprend en particulier les hémangiomes infantiles et les hémangiomes congénitaux. Les hémangiomes infantiles sont les tumeurs vasculaires les plus fréquentes, et se voient chez le nourrisson.
- Le second groupe est celui des malformations vasculaires. Dans cette situation, les vaisseaux ne se multiplient pas, mais leur structure est anormale dès le départ, par exemple avec un diamètre augmenté. Il s’agit d’anomalies présentes dès la naissance, même si elles ne sont pas toujours visibles immédiatement. On distingue plusieurs types de malformations vasculaires : les malformations capillaires (aussi appelées angiomes plans), les malformations lymphatiques (ou lymphangiomes), les malformations veineuses et les malformations artério-veineuses.
1. Hémangiome infantile
L’hémangiome infantile, qu'est-ce-que c'est ?
Les hémangiomes infantiles sont les anomalies vasculaires les plus fréquentes chez le nourrisson. Ce sont des amas de petits vaisseaux sanguins qui se développent de façon excessive pendant un certain temps, puis finissent le plus souvent par régresser spontanément.
Ce ne sont pas des lésions cancéreuses, et dans la grande majorité des cas ne comportent pas de risque.
Les hémangiomes infantiles, parfois appelés « angiomes tubéreux » ou « angiomes fraise » en raison de leur aspect, concernent environ 5 à 10 % des nourrissons.
Ils apparaissent le plus souvent après la naissance, au cours des premières semaines de vie, alors que la peau était normale à la naissance (parfois une petite tache rouge, plane et discrète, peut être visible au départ).
Ils sont plus fréquents chez les filles, chez les nourrissons de petit poids de naissance, en cas de grossesse multiple (avec une augmentation de la fréquence chez les jumeaux), ou en cas de complications pendant la grossesse, comme une hypertension artérielle gravidique (tension élevée pendant la grossesse) ou un décollement placentaire. Les nourrissons prématurés présentent aussi un risque plus élevé de développer un hémangiome infantile.
Sur le plan clinique, on distingue trois grands types d’hémangiomes infantiles.
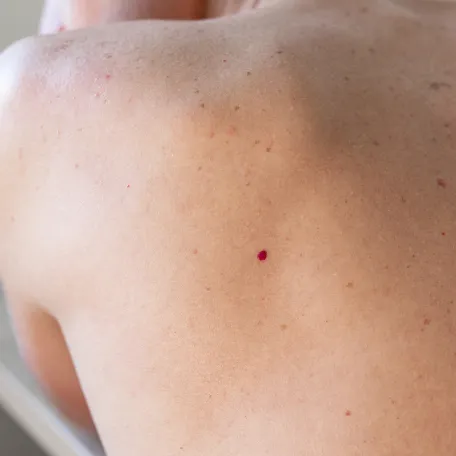
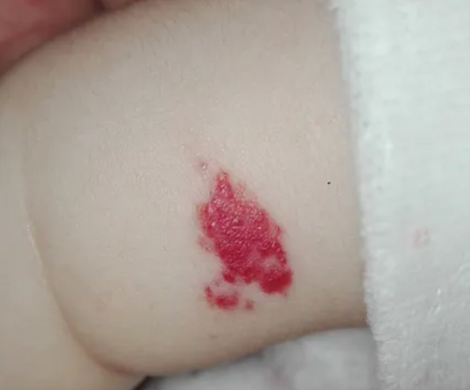
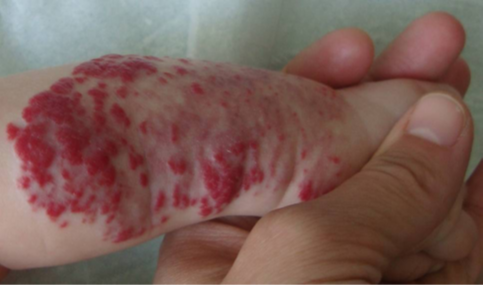
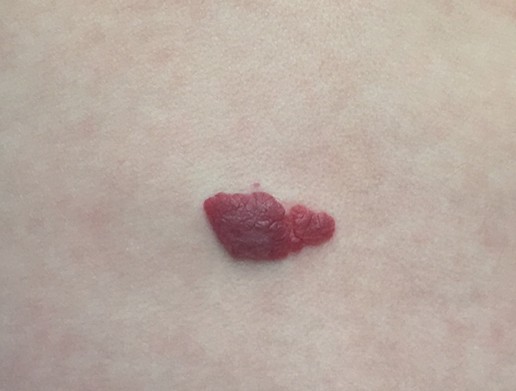
- Les hémangiomes infantiles superficiels qui sont les plus connus : ils sont de couleur rouge vif, bien délimités, avec une surface lisse ou parfois un peu granuleuse (aspect ‘en fraise’).
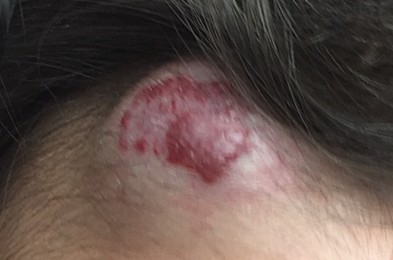
- Les hémangiomes infantiles superficiels sous-cutanés : ils sont situés plus en profondeur sous la peau et se manifestent par une tuméfaction (un gonflement) saillante, de couleur bleutée, ferme mais souple au toucher.
- Les formes mixtes associent à la fois une partie visible en surface et une composante plus profonde sous la peau.
Hémangiome infantile superficiel
.
Hémangiome infantile superficiel
.
Hémangiome infantile superficiel
.
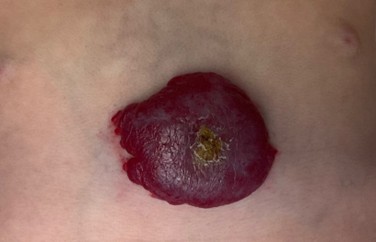
Hémangiome infantile superficiel tubéreux
.
Hémangiome infantile profond
.
Hémangiome infantile mixte
.
La taille d’un hémangiome infantile peut varier de quelques millimètres à plusieurs centimètres. Dans certains cas, une grande partie d’un membre ou du visage peut être atteinte. Certains enfants peuvent également présenter plusieurs hémangiomes en même temps.
La localisation de l’hémangiome infantile est un élément important, car elle aide le médecin à évaluer le risque de complications et à décider s’il est nécessaire de proposer des examens complémentaires ou un suivi spécialisé.
Certaines localisations nécessitent une attention particulière, car elles peuvent être associées à un risque plus élevé de gêne ou de complications. C’est notamment le cas des hémangiomes infantiles situés autour de l’œil, qui peuvent gêner la vision, du nez ou de la bouche, pouvant entraîner une gêne pour l’alimentation ou la respiration, de la région anogénitale, ou encore d’un membre lorsqu’ils peuvent gêner la marche ou l’utilisation de la main.
Les hémangiomes infantiles qui occupent une grande zone du corps, appelés hémangiomes infantiles segmentaires, peuvent plus rarement être associés à d’autres anomalies vasculaires ou malformatives plus complexes. Ces situations restent rares, mais elles expliquent pourquoi une évaluation médicale adaptée est parfois nécessaire en fonction de la localisation de l’hémangiome.
L’évolution des hémangiomes infantiles se fait classiquement en trois phases.
- Il existe d’abord une phase de croissance, au cours de laquelle l’hémangiome augmente de taille. Cette phase débute souvent après quelques semaines de vie et peut durer de quelques semaines à cinq ou six mois, parfois jusqu’à un an dans certaines formes plus importantes. La croissance est en général plus rapide au cours des premiers mois, en effet la plupart des hémangiomes infantiles grandissent surtout au début : environ 80 % de leur croissance est déjà faite avant l’âge de 3 mois1.
- Ensuite, l’hémangiome infantile se stabilise,
- Enfin il entre dans une phase de diminution de volume spontanée. Progressivement, il pâlit, devient moins ferme et diminue de taille.
Dans environ 90 % des cas, les hémangiomes infantiles ont complètement régressé à l’âge de 4 ans 2,3.
Il persiste toutefois fréquemment des modifications de la peau à l’endroit de l’hémangiome, comme une peau plus fine, une poche ou une légère décoloration.
Dans environ 30 % des cas, des séquelles plus visibles persistent après la phase de régression. Elles peuvent être principalement esthétiques, mais parfois aussi fonctionnelles selon la localisation. Lorsque ces séquelles sont jugées gênantes, des traitements peuvent être proposés, comme le laser et/ou la chirurgie.
Faut-il traiter un hémangiome infantile ?
Aucun traitement n'est nécessaire pour la plupart des hémangiomes infantiles.
Dans la grande majorité des cas, aucun traitement n’est nécessaire, car la plupart des hémangiomes infantiles régresse spontanément avec le temps. Une simple surveillance médicale est alors suffisante. Cette attitude dite ‘attentiste’ est la plus fréquente puisqu’environ 80 % des hémangiomes du nourrisson ne sont pas traités.
En revanche, certains hémangiomes infantiles peuvent se compliquer ou entraîner une gêne importante, et une consultation spécialisée auprès d’un dermatologue pédiatre ou d’une équipe expérimentée est alors nécessaire. C’est notamment le cas si l’hémangiome infantile s’ulcère (la peau devient fragile et se creuse), ce qui est douloureux pour l’enfant et peut favoriser des infections. Une prise en charge est également indiquée lorsque l’hémangiome infantile augmente rapidement de volume pendant sa phase de croissance et qu’il est situé dans une zone sensible, par exemple sur le nez, une lèvre, à proximité d’un œil ou des voies respiratoires, avec un risque de déformation, de compression ou de gêne de la vision, de l’alimentation ou de la respiration.
Une consultation spécialisée auprès d’un dermatologue pédiatre ou d’une équipe expérimentée est ainsi nécessaire si :
- L’hémangiome infantile s'ulcère, ce qui est douloureux pour l'enfant et peut favoriser des infections
- L'hémangiome infantile prend un volume important lors de sa phase de croissance et qu'il se situe, par exemple, sur le nez, une lèvre, à proximité d’un œil ou des voies respiratoires, avec un risque de déformation, de compression ou de gêne de la vision, de l’alimentation ou de la respiration.
En fonction du type d’hémangiome infantile, de sa localisation et de l’âge de l’enfant, différents traitements peuvent être proposés.
Aujourd’hui, les bêtabloquants constituent le traitement de référence des hémangiomes infantiles nécessitant un traitement, les autres options (laser, chirurgie) étant réservées à des situations particulières ou utilisées en complément.
Les bêtabloquants et les hémangiomes infantiles
Lorsque l’hémangiome infantile est volumineux, ulcéré ou à risque de séquelles, un traitement peut être proposé. Le propranolol par voie orale est actuellement le traitement de première intention dans ces formes, car il permet de freiner rapidement la croissance de l’hémangiome et d’en favoriser la régression.
- Avant de débuter un traitement par propranolol, un examen médical complet est toujours réalisé, avec la mesure du pouls et de la tension artérielle, afin de s’assurer qu’il n’existe pas de contre-indication. Un avis cardiologique sera demandé si l’enfant présente des antécédents cardiaques ou des anomalies à l’examen clinique.
- En France, en 2026, le traitement est le plus souvent initié en milieu hospitalier, en particulier dans certaines situations (nourrisson très jeune, prématurité, faible poids, comorbidités). Cette initiation à l’hôpital permet une surveillance rapprochée afin de s’assurer de la bonne tolérance du traitement.
- La dose de propranolol est adaptée au poids de l’enfant et augmentée progressivement.
- Les premiers effets sont souvent rapides, avec un ramollissement et une diminution de la coloration de l’hémangiome en quelques jours, puis une régression progressive sur plusieurs mois. Le traitement est en général poursuivi jusqu’à la fin de la phase de croissance de l’hémangiome, le plus souvent autour de l’âge d’un an, parfois plus longtemps selon la situation.
- Le traitement par propranolol est globalement bien toléré. Une surveillance régulière est assurée par le médecin. Certains effets secondaires peuvent survenir, le plus souvent modérés, comme des mains ou des pieds froids ou des troubles du sommeil. Le propranolol peut favoriser des baisses de sucre dans le sang, ce qui explique l’importance de donner le médicament pendant ou juste après un repas et de respecter des horaires réguliers d’alimentation. Plus rarement, il peut révéler une tendance à l’asthme, avec des sifflements respiratoires, notamment lors d’infections respiratoires, ce qui nécessite alors une réévaluation du traitement.
Pour les hémangiomes infantiles superficiels de petite taille, un traitement par bêtabloquant local peut parfois être proposé. Il s’agit le plus souvent de timolol sous forme de gel ou de solution appliquée directement sur la peau (c’est une prescription courante bien que ce traitement n’ait pas d’autorisation sur le marché officielle dans cette indication). Ce traitement local est réservé aux formes peu profondes et limitées. Il présente l’avantage de limiter le passage du médicament dans le reste de l’organisme, avec un risque d’effets secondaires généraux très faible, tout en nécessitant une surveillance médicale.
Il est également possible que le dermatologue propose de participer à une étude clinique. Ces études permettent d’améliorer les connaissances et d’optimiser les traitements proposés aux enfants.
2. Hémangiome congénital
Les hémangiomes congénitaux, qu’est-ce que c’est ?
Les hémangiomes congénitaux sont des anomalies vasculaires rares, présentes dès la naissance, contrairement aux hémangiomes infantiles qui apparaissent le plus souvent après quelques semaines de vie.
Ils résultent d’une prolifération excessive et localisée de vaisseaux sanguins de la peau pendant la vie in utero. Ces lésions sont bénignes, c’est-à-dire non cancéreuses. Leur évolution est différente de celle des hémangiomes infantiles.
On distingue principalement deux types d’hémangiomes congénitaux.
- Les hémangiomes congénitaux à involution rapide, appelés RICH (Rapidly Involuting Congenital Hemangioma). Leur taille est maximale dès la naissance, et ne présentent pas de phase de croissance, à l’inverse des hémangiomes infantiles. Ils régressent ensuite spontanément et rapidement après la naissance, le plus souvent au cours de la première année de vie.
- Les hémangiomes congénitaux non involutifs, appelés NICH (Non-Involuting Congenital Hemangioma), sont généralement peu volumineux mais, contrairement aux RICH, ne régressent pas et restent stables tout au long de la vie.
Les hémangiomes congénitaux sont le plus souvent uniques et siègent fréquemment sur les membres, le tronc ou la tête.
Quels examens sont nécessaires ?
Le diagnostic d’un hémangiome congénital repose avant tout sur l’examen clinique et sur le fait que la lésion est déjà présente à la naissance. Le médecin s’appuie sur l’aspect, la localisation et l’évolution dans le temps pour orienter le diagnostic.
Une échographie Doppler ou une IRM peut être réalisée afin d’analyser la structure des vaisseaux et la circulation sanguine à l’intérieur de la lésion. Cet examen est indolore et permet de confirmer la nature vasculaire de l’hémangiome.
Quels sont les traitements ?
La prise en charge des hémangiomes congénitaux dépend principalement de leur type (RICH ou NICH), de leur taille, de leur localisation et de la gêne qu’ils peuvent entraîner.
- Dans le cas des RICH, aucun traitement n’est généralement nécessaire. Une surveillance médicale est suffisante, car la lésion régresse spontanément. Après la régression, il peut persister des modifications de la peau, comme une peau plus fine, un aspect fripé ou une cicatrice, qui peuvent parfois justifier un traitement secondaire à visée esthétique.
- Les NICH, en revanche, ne régressent pas spontanément. Lorsqu’ils ne provoquent ni douleur, ni gêne fonctionnelle ou esthétique importante, une simple surveillance peut être proposée. En revanche, si le NICH est volumineux, gênant ou responsable d’un retentissement esthétique important, un traitement peut être discuté.
- Contrairement aux hémangiomes infantiles, les hémangiomes congénitaux ne répondent pas au traitement par bêtabloquants, qui n’est donc pas indiqué dans cette situation.
- Lorsqu’un traitement est envisagé, il repose le plus souvent sur la chirurgie, réalisée à distance de la naissance lorsque cela est possible, parfois associée à des techniques de laser.
3. Angiome plan ou malformation capillaire
Qu’est-ce qu’une malformation capillaire, couramment appelée angiome plan ?
Ce que l’on appelle couramment un angiome plan est aujourd’hui nommé, en pratique médicale, malformation capillaire. Il est aussi connu sous les noms de ‘tache de vin’.
Il s’agit d’une anomalie vasculaire superficielle présente dès la naissance. Les petits vaisseaux sanguins de la peau sont anormalement formés : ils sont plus larges que la normale et restent visibles à travers la peau.
L’angiome plan concerne environ 3 à 5 bébés sur 1 000 à la naissance. Il touche aussi bien les filles que les garçons.
Il est le plus souvent situé au niveau du visage et du cou, mais peut également atteindre les bras ou les jambes.
Cette anomalie est bénigne, c’est-à-dire ni dangereuse ni cancéreuse.
Contrairement aux hémangiomes infantiles, l’angiome plan ne disparaît pas spontanément avec le temps. Il persiste toute la vie. En revanche, il ne s’étend pas à de nouvelles zones, mais il grandit proportionnellement avec la croissance de l’enfant.
Comment se manifeste un angiome plan ?
L’angiome plan se manifeste par une tache plane, bien limitée, de couleur rose, rouge ou violacée.
Avec le temps, notamment à l’âge adulte, la tache peut devenir plus foncée, plus épaisse ou prendre un aspect légèrement granuleux. Sa couleur peut aussi varier selon la température, les émotions ou l’effort.
L’angiome plan est indolore. Dans la grande majorité des cas, la gêne est exclusivement esthétique.
Il est important de ne pas confondre l’angiome plan avec l’angiome en flammèche, très fréquent chez le nourrisson et complètement bénin. L’angiome en flammèche correspond à une coloration rosée à rouge clair à bords émiettés, souvent située sur le front et sur les paupières supérieures mais les atteignant de manière symétrique et sans vraie continuité entre la lésions du front et celle des paupières. Il a souvent une forme de triangle inversé (base vers en haut et point vers le bas). On le retrouve également au niveau de la nuque. Il s’agit d’une marque très fréquente chez les bébés, transitoire, qui s’estompe puis disparaît spontanément en quelques mois ou quelques années, sans conséquence.
Quelles sont les localisations à surveiller ?
Certaines localisations d’angiome plan nécessitent une attention particulière.
- Un angiome plan situé sur le visage, le plus souvent d’un seul côté (asymétrique), touchant le front et la paupière supérieure, peut être associé au syndrome de Sturge-Weber. Ce syndrome associe un angiome plan du visage à une atteinte des vaisseaux des méninges (les membranes qui entourent le cerveau) et parfois à un glaucome, c’est-à-dire une augmentation de la pression à l’intérieur de l’œil. Dans ce cas, un suivi ophtalmologique et parfois neurologique est nécessaire.
- Un angiome plan situé sur un membre, en particulier le membre inférieur, peut être associé à une croissance anormale du membre concerné, en longueur et en diamètre. Cette asymétrie peut nécessiter un suivi orthopédique spécialisé, surtout pendant la croissance.
Faut-il réaliser des examens ?
Dans la majorité des cas, aucun examen complémentaire n’est nécessaire.
Des examens peuvent toutefois être proposés en fonction de la localisation de l’angiome plan, par exemple un examen ophtalmologique et une IRM cérébrale en cas d’atteinte du visage dans une zone à risque. Lorsque l’angiome plan est situé sur un membre, des examens d’imagerie des vaisseaux du membre concerné peuvent parfois être proposés, en particulier si le médecin suspecte une anomalie associée
Quel est le traitement de l’angiome plan ?
Le traitement de référence de l’angiome plan est le laser à colorant pulsé. Ce laser agit en ciblant les petits vaisseaux anormaux responsables de la coloration de la tâche, sans abîmer la peau environnante.
Pour obtenir un résultat satisfaisant, plusieurs séances sont nécessaires, en moyenne quatre à six, parfois davantage selon l’étendue et l’intensité de la lésion.
Dans de nombreux cas, le laser à colorant pulsé ne permet pas de faire disparaître complètement l’angiome plan, mais de l’éclaircir de façon importante. Les résultats sont généralement meilleurs lorsque le traitement est débuté tôt, même si le laser peut être proposé à tout âge. Le traitement est avant tout esthétique et vise à améliorer l’aspect de la peau et la qualité de vie.
4. Malformation lymphatique
Qu’est-ce qu’une malformation lymphatique (lymphangiome) ?
Un lymphangiome est une malformation des vaisseaux lymphatiques. Les vaisseaux lymphatiques font partie du système lymphatique, qui participe à la défense de l’organisme et à la circulation des liquides dans le corps.
Dans le cas d’un lymphangiome, ces vaisseaux sont malformés dès la vie avant la naissance. Il s’agit d’une anomalie bénigne, c’est-à-dire ni cancéreuse ni contagieuse.
Les lymphangiomes sont le plus souvent présents dès la naissance ou apparaissent dans les premières années de vie, même s’ils peuvent parfois passer inaperçus au début.
On distingue principalement deux grands types de lymphangiomes.
- Les lymphangiomes macrokystiques : ils se présentent comme des grosses poches ou kystes remplis de liquide, situés sous la peau. Ils sont généralement mous au toucher. Les localisations les plus fréquentes sont le visage, le cou, les aisselles ou le thorax.
Selon leur taille et leur emplacement, ils peuvent parfois comprimer les organes voisins. Des épisodes inflammatoires douloureux peuvent parfois survenir, avec une augmentation brutale du volume, une rougeur et parfois de la fièvre.
- Les lymphangiomes microkystiques : ils se manifestent souvent à la surface de la peau par de petites vésicules, ressemblant à de minuscules cloques. Leur contenu peut être clair ou parfois légèrement sanglant. Ces vésicules peuvent être isolées ou regroupées sur une zone plus étendue.
Dans certains cas, l’aspect est moins typique : on peut simplement observer un épaississement ou un gonflement de la peau, sans lésions visibles.
Quels examens sont nécessaires en cas de malformation lymphatique ?
Une malformation lymphatique peut parfois être repéré dès la grossesse, lors d’une échographie prénatale.
Après la naissance, le diagnostic repose avant tout sur l’examen clinique.
Selon la situation, des examens d’imagerie peuvent être demandés pour mieux visualiser la malformation, préciser sa taille et son extension :
- Une échographie, parfois associée à un Doppler
- Une IRM, notamment lorsque la malformation est profonde ou étendue.
Ces examens sont indolores et permettent d’adapter au mieux la prise en charge.
Quel est le traitement des malformations lymphatiques ?
Le traitement dépend du type de malformation lymphatique, de sa taille, de sa localisation et de la gêne qu’il entraîne.
- Pour les formes macrokystiques, le traitement de première intention est aujourd’hui la sclérothérapie. Cette technique consiste à injecter un produit directement dans les kystes afin de les faire diminuer de volume.
Le sirolimus (inhibiteur de mTOR qui inhibe la prolifération des vaisseaux lymphatiques) peut être associé à la sclérothérapie ou prescrit seul et permet de diminuer les poches lymphatiques. Cette prescription se fait par les équipes expertes pluridisciplinaires dans des centres de référence. La chirurgie peut être envisagée dans certaines situations particulières, mais elle n’est pas toujours possible ni nécessaire (récidive fréquente après chirurgie).
- Pour les formes microkystiques, une abstention thérapeutique (c’est-à-dire une simple surveillance) peut être tout à fait raisonnable lorsque les symptômes sont limités. La chirurgie n’est pas recommandée (récidive fréquente) ni le laser (douloureux et efficacité incomplète).
Une étude a évalué récemment le sirolimus sous forme de crème. Les résultats seront publiés prochainement.
- En cas de poussée inflammatoire douloureuse, un traitement médical est généralement prescrit par antibiotiques et corticoïdes. Ces traitements visent à calmer l’inflammation, mais ne font pas disparaître la malformation.
5. Les malformations veineuses
Elles sont plus rares. Elles se présentent comme une ou plusieurs tuméfactions bleutées ressemblant à des varices qui augmentent de volume lors des efforts, en position debout prolongée ou par temps chaud.
Elles peuvent devenir douloureuses lors d’épisodes de thromboses, qui correspondent à la formation d’un petit caillot dans une veine au sein de la malformation. Ces thromboses sont superficielles sans risque de phlébite ou d’embolie pulmonaire.
Le traitement des malformations veineuses est souvent complexe. Lorsqu’elles sont petites, bien limitées et accessibles, un traitement chirurgical complet peut parfois être proposé. Le plus souvent, cependant, il n’est pas possible de les faire disparaître totalement.
La prise en charge repose alors sur des traitements partiels, comme la sclérothérapie (injection d’un produit dans la veine pour la faire se rétracter) et/ou des interventions chirurgicales limitées, dans le but de diminuer le volume, la douleur ou la gêne.
6. Les malformations artérioveineuses
Les malformations artério-veineuses sont les anomalies vasculaires les plus rares et les plus complexes. Elles correspondent à une connexion anormale entre des artères et des veines, sans passer par les petits vaisseaux intermédiaires qui existent normalement pour relier ces deux types de vaisseaux sanguins. Cela entraîne une circulation du sang trop rapide et mal régulée dans la zone concernée.
Chez l’enfant, ces malformations peuvent rester longtemps peu visibles. On parle alors d’un stade dit « dormant ». À ce stade, elles peuvent parfois ressembler à un angiome plan, c’est-à-dire à une tache rose, rouge ou violacée, plane ou avec très peu de relief.
Certains signes doivent toutefois alerter. Il peut s’agir d’une chaleur anormale au niveau de la lésion, ou d’une augmentation de taille inhabituelle. Contrairement à l’angiome plan, qui grandit simplement en même temps que l’enfant et de façon proportionnelle à sa croissance, la malformation artério-veineuse évolue par elle-même : elle peut s’étendre plus rapidement, devenir plus épaisse ou plus étendue que ce que l’on attendrait avec la croissance normale de l’enfant.
C’est le plus souvent à la puberté que la malformation artério-veineuse évolue plus rapidement et devient plus visible. Son évolution au cours de la vie est imprévisible, notamment pendant les grossesses. Elle peut progressivement envahir les tissus voisins, comme la peau, les muscles et parfois même les os.
Le traitement est difficile et nécessite une prise en charge dans des centres spécialisés. Il repose sur le travail d’une équipe multidisciplinaire expérimentée, associant dermatologues, radiologues interventionnels et chirurgiens, afin d’adapter au mieux la stratégie thérapeutique à chaque situation.
7. Quelques anomalies vasculaires fréquentes et banales
- Les angiomes stellaires sont de petites anomalies ou les vaisseaux sont disposés en étoile. Ils sont fréquents à la partie supérieure du corps, en particulier chez les enfants et les femmes, surtout lors de la grossesse. S'ils deviennent gênants esthétiquement et qu’ils ne disparaissent pas spontanément, un traitement par laser ou électrocoagulation peut être proposé.
- Les botriomycomes, aussi appelés bourgeons charnus, apparaissent souvent sur des zones fragilisées ou traumatisées (ongle incarné, petite blessure etc.), mais peuvent aussi survenir spontanément. Ils ressemblent à une petite boule rouge, parfois très vascularisée, qui peut saigner facilement. Leur traitement repose sur l’ablation et/ou l’électrocoagulation.
- Les lacs veineux des lèvres sont fréquents après 60 ans. Ils ressemblent à une petite varice bleu violacé sur la lèvre. Ils sont bénins et ne nécessite pas de prise en charge. En cas de gêne esthétique importante, un traitement par laser peut être proposé s’ils sont petits, sinon une ablation chirurgicale peut être discutée.
- Les angiomes rubis aussi appelés taches rubis, sont très fréquents après 40 à 50 ans. Ce sont de petites excroissances rouges, de quelques millimètres, sans gravité. Ils augmentent souvent en nombre avec l’âge. Un traitement par laser est possible pour des raisons esthétiques.
- Les angiokératomes sont plus foncés que les angiomes rubis. Ils apparaissent sous forme de petites lésions rouge foncées, violacées ou brunâtres. Lorsqu’ils apparaissent après 60 ans, notamment sur les organes génitaux, ils sont le plus souvent bénins et sans conséquence.
En revanche, l’apparition d’angiokératomes multiples et précoces, avant l’âge de 20 ans, doit faire évoquer une maladie génétique rare appelée maladie de Fabry, qui nécessite un avis médical spécialisé.
info Télécharger la fiche
Références
- for the Hemangioma Investigator Group, Growth Characteristics of Infantile Hemangiomas: Implications for Management, 2008, https://doi.org/10.1542/peds.2007-2767
- Christine Léauté-Labrèze, John I Harper, Peter H Hoeger, Infantile haemangioma, 2017, https://doi.org/10.1016/s0140-6736(16)00645-0
- Constantijn G. Bauland, Thomas H. Lüning, Jeroen M. Smit, Clark J. Zeebregts, Paul H. M. Spauwen, Untreated Hemangiomas: Growth Pattern and Residual Lesions, 2011, https://doi.org/10.1097/prs.0b013e318208d2ac


